


Researchers at NYU Shanghai have proposed a new approach for constructing theoretical chemical models that can be used to compute charge and energy transfer rates and pathways in different organic semiconductor materials, and provide benchmarks for examining the performance of quantum dynamic methods. The findings were published in The Journal of Chemical Physics, a leading journal in the field. NYU Shanghai Assistant Professor of Chemistry Sun Xiang conceived the study and supervised the research. His group members, Hu Zhubin, Postdoctoral Fellow of Chemistry, and PhD student Dominikus Brian, were key researchers.
A central topic of theoretical and computational chemistry focuses on analyzing the physical movements of atoms and molecules using quantum chemistry and molecular dynamics simulations. “When atoms and molecules absorb external energy, such as sunlight, the electric charges inside them flow, and thus electric currents are created. The process is called ‘photoinduced charge transfer.’ By simulating this process, we can compute the charge transfer pathways in a condensed-phase environment, which lays the theoretical groundwork for the study of solar energy conversion efficiency and may contribute to the discovery of more efficient artificial energy-conversion materials,” said Sun.
Molecules have different electronic states. “For example, there’s a ‘ground state’, under which the electrons in the molecules are at their lowest energy. Electrons can also get ‘excited’ when being stimulated by light, and they are called the ‘excited states’,” said Sun. Previous studies in the field had constructed matured models to simulate the photoinduced charge transfer process involving two electronic states. However, there was no consistent approach to construct models for the analysis in multi-state scenarios.
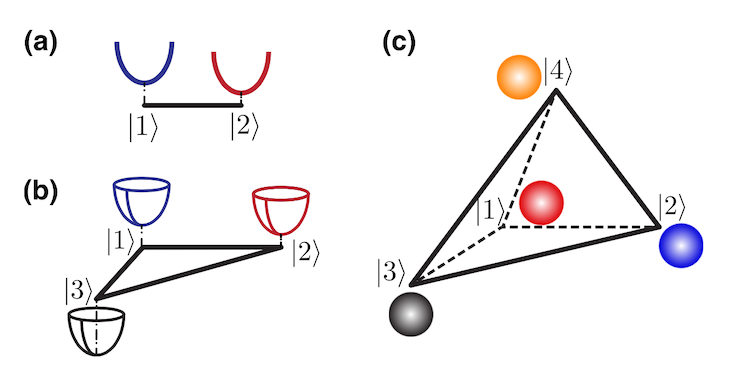
Illustration of the potential energy surfaces in the two-state model (a), three- state model (b), and four-state model (c).
In their study, Sun’s team successfully proposed an approach for building harmonic models for photoinduced charge transfer under an arbitrary number of electronic states by conducting theoretical calculations with resources from NYU Shanghai’s High Performance Computing. “The proposed models provide a general and flexible framework for simulating nonadiabatic dynamics in complex systems. More importantly, they can serve as benchmarks for examining the performance of other dynamical methods, thus helping the selection of appropriate methods for the studies of different materials,” Sun said.
Computational chemistry is one of NYU Shanghai’s growing research areas. “Our group is small but full of passion and dedication. With fewer than 10 members in the group, we’re able to provide individualized mentorship according to each student’s situation and need,” Sun said. “The NYU-ECNU Center for Computational Chemistry at NYU Shanghai is in a unique position to interact with other academic institutions, with close ties to New York University’s global network, as well as major universities and research institutions in China and abroad. Through frequent communication and collaboration with domestic and international researchers, universities, and institutes, we’re able to closely follow the most updated research trend and development.”

Sun Xiang’s Group
After finishing his research at NYU Shanghai, Hu, the first author of the paper, recently joined East China Normal University as a full-time researcher. Brian, the second author, is continuing his PhD study in Sun’s group. The research received funding from the National Natural Science Foundation of China.